“蛋白质”浙大团队用深度学习方法进行高效、准确的大型文库配体对接,助力药物开发
今天,很高兴为大家分享来自机器之心Pro的浙大团队用深度学习方法进行高效、准确的大型文库配体对接,助力药物开发,如果您对浙大团队用深度学习方法进行高效、准确的大型文库配体对接,助力药物开发感兴趣,请往下看。
第一时间掌握
 编辑 | 萝卜皮
编辑 | 萝卜皮配体对接是基于结构的药物发现虚拟筛选的核心技术之一。然而,传统的对接工具和现有的深度学习工具可能在速度、位姿(pose)质量和结合亲和力准确性方面受到性能限制。
浙江大学团队提出了 KarmaDock,一种用于配体对接的深度学习方法,它集成了对接加速、结合位姿生成和校正以及结合强度估计的功能。
模型由三部分组成:(1)蛋白质和配体的编码器,用于学习分子内相互作用的表示;(2)E(n) 等变图神经网络,具有自注意力机制,可根据蛋白质-配体和分子内相互作用更新配体位姿,然后进行后处理以确保化学上合理的结构;(3)用于对结合强度进行评分的混合物密度网络。
KarmaDock 在四个基准数据集上进行了验证,并在现实世界的虚拟筛选项目中进行了测试,该项目成功识别了经过实验验证的白细胞酪氨酸激酶(LTK)活性抑制剂。
该研究以「Efficient and accurate large library ligand docking with KarmaDock」为题,于 2023 年 9 月 21 日《Nature Computational Science》。

配体对接概述
配体对接是基于结构的虚拟筛选(VS)的核心任务之一,在蛋白质-配体(PL)结合位姿生成、PL结合亲和力预测、PL结合位姿选择和 VS 中发挥着关键作用。一般来说,传统对接程序(例如 AutoDock 4、AutoDock Vina、LeDock、Glide 和 GOLD)利用启发式搜索算法来探索一系列可能的配体构象,以及带有简化术语的评分函数 (SF),用于配体位姿选择和 PL 结合强度估计。这种简化提高了大规模 VS 的效率,但这是以精度下降为代价的。随着化合物库规模的不断增大,对超大型 VS 更快方法的需求引起了学界对一系列加速方法的开发,例如 QuickVina 2 和 AutoDock GPU,以及用于预测结合亲和力和生成结合位姿的深度学习 (DL) 技术。
随着 AlphaFold2 在从蛋白质序列预测原子水平上的蛋白质结构方面取得了显著成就,越来越多的研究人员开始应用 DL 算法来预测配体构象和 PL 结合位姿。与传统的配体对接方法不同,利用深度学习算法(尤其是图神经网络,GNN)预测 PL 构象可以加速对接过程并提高对接精度。
已有方法的局限性
基于深度学习的位姿生成模型 (PGM) 不是搜索和评分,而是通过以下三种方法之一生成结合位姿:(1)使用梯度下降(例如 TankBind)预测 PL 距离矩阵并生成 PL 结合位姿;(2)实现 E(n) 等变图神经网络(EGNN)层来预测每次消息传递迭代中配体原子的运动和方向(例如,EquiBind15 和 E3Bind);(3)利用去噪扩散概率模型来预测配体的平移、旋转和扭转,类似于传统的对接工具(例如 DiffDock)。方法(1)和(3)都能够生成具有化学上合理的局部结构的构象,尽管效率有限。相反,基于 EGNN 的模型表现出卓越的速度,但在生成具有化学上合理的键长和键角的构象方面面临挑战。
尽管这些 DL 模型(EquiBind 除外)在对接位姿生成方面取得了实质性改进,但它们在 PL 复合体的结合位点(或口袋)未知的盲对接场景中最为有效。然而,在 VS 实践中,通常通过事先实验已知或确定结合位点,更频繁地使用口袋给定的配体对接,而传统的对接工具主要是为此目的而设计的。
为了解决这个问题,引入了 LigPose 作为一种口袋引导方法,利用具有自注意力的 EGNN 来生成 PL 结合位姿并预测其结合亲和力。LigPose 实现了令人印象深刻的高对接成功率和低评分错误,与传统工具相比最多可加速 265 倍。然而,与传统的对接方法不同,预测的配体构象可能不符合键长和角度的物理规则,因为在原子运动预测期间没有施加几何约束。
尽管对接算法可以产生可接受的对接构象,但结合亲和力预测(也称为化合物-蛋白质相互作用,CPI)的准确性通常不能令人满意,这导致了使用各种 DL 算法的深度学习评分函数(DLSF)模型的开发。在 DLSF 中,RTMScore使用混合密度网络(MDN)来学习每个 PL 节点的距离分布,并将结合强度确定为 PL 节点的概率之和,实现了令人印象深刻的筛选能力和对接能力。尽管 DLSF 在结合亲和力预测方面可以优于传统 SF,但其准确性很大程度上取决于配体结合位姿的质量。
一种称为KarmaDock的DL方法
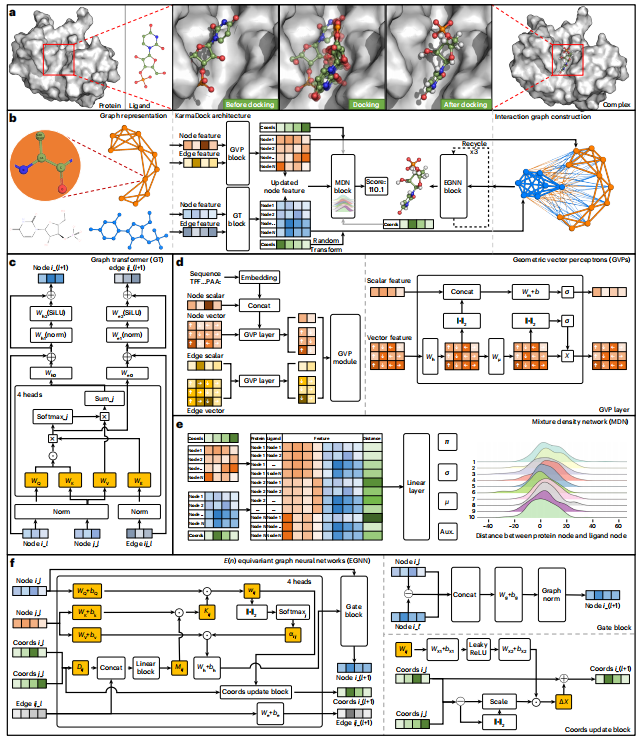 图示:KarmaDock 的工作流程。(来源:论文)
图示:KarmaDock 的工作流程。(来源:论文)在最新的研究中,浙江大学的研究人员提出了一种称为 KarmaDock 的深度学习方法,用于口袋引导配体对接,该方法可以快速、高精度地生成结合位姿并预测结合强度。KarmaDock 框架实现了由两个编码器 [graph transformer (GT) 和几何向量感知器 (GVP)]、一个用于评分的 MDN 块和一个用于对接的 EGNN 块组成的 GNN 架构。
该方法的创新点如下:(1)基于残基而不是原子来表征蛋白质,从而编码几何特征并降低计算成本;(2)MDN 块学习到的每个蛋白质与配体节点之间的最小距离的概率分布可以向共享编码器引入距离归纳偏差,从而有助于指导位姿生成的学习;(3)设计两个编码器分别接收距离感应偏差并学习蛋白质和配体的分子内相互作用;(4)实现全连接交互图与基于自注意力的 EGNN 相结合,以实现快速对接;(5)采用两种后处理方法,保证生成的构象在键长和角度方面合理。
KarmaDock 在四个配体对接基准数据集(PDBBind 2020 版、APObind、CASF 2016 和 DEKOIS 2.0 版,用于位姿生成、位姿选择和 VS 任务)上进行了评估。它还已应用于基于 ChemDiv 和 Specs 数据库的现实 VS 中,用于识别白细胞酪氨酸激酶(LTK)抑制剂。对于位姿生成,KarmaDock 在准确性和速度方面优于所有传统对接工具,生成配体位姿的对接成功率为 89.1%,每个复合物的速度为 0.017 s,并再现了大多数相互作用模式。
对于位姿选择,KarmaDock 的成功率达到 95.6%,在所有方法中排名第二。就 VS 而言,对接精度、速度和结合强度都很重要,KarmaDock 在 CASF 2016 和 DEKOIS 2.0 版数据集上的平均富集因子分别为 23.4 和 16.3,高于传统对接工具所达到的水平。此外,KarmaDock 能够在单个 Tesla V100 上在 8.4 小时(AutoDock GPU 为 57.1 天)内筛选 177 万种化合物,并成功发现了经过实验验证的具有亚微活性的 LTK 抑制剂。这些结果证明了 KarmaDock 的卓越性能,并表明它是超大型 VS 的一个有前途的工具。
结语
在这项研究中,研究人员努力实施后处理技术,例如 FF 优化和 RDkit 构象比对,作为对预测配体构象中检测到的键长和角度不准确的纠正措施。
值得注意的是,这些后处理方法确实增强了构象合理性,尽管代价是对接精度下降,在两者之间取得平衡是一项具有挑战性的任务。实用的解决方案似乎是对化合物库进行分层筛选,其中筛选过程最初绕过构象理性,然后对得分最高的复合物进行 FF 优化。进一步的实验和深入的实验验证可以证实该解决方案并拓宽KarmaDock的应用范围。
然而,值得承认的是 KarmaDock 的固有局限性,它是一种半灵活的对接工具,对蛋白质结构变化视而不见。此属性或许可以解释 APObind 性能明显下降的原因。因此,该团队的前瞻性策略是将蛋白质结构可变性纳入 KarmaDock 的范围内,最终目标是将其转变为完全灵活的对接工具。
论文链接:https://www.nature.com/articles/s43588-023-00511-5
好了,关于浙大团队用深度学习方法进行高效、准确的大型文库配体对接,助力药物开发就讲到这。
版权及免责声明:凡本网所属版权作品,转载时须获得授权并注明来源“科技金融网”,违者本网将保留追究其相关法律责任的权力。凡转载文章,不代表本网观点和立场,如有侵权,请联系我们删除。
相关文章
- “考生”下半年自考即将开始 省考试院发出温馨提示
- “父亲”父亲的眼神杀
- “这是”自内耗到自洽
- “大桥”G3铜陵长江公铁大桥先导索过江
- “某甲”父亲被羁押继母要离婚,未成年女儿谁来抚养?法官多方努力,难题解决了!
- “亚马逊”哪些以色列芯片公司已被美国企业收购?
- “结构”结核杆菌致病机制获揭示
- “装修”装修公司老板明知公司亏损仍吸引客户签合同,骗取上百人700余万,被判11年
- “射电”穿越80亿光年的快速射电暴源于一场“星系交通事故”
- “必胜客”必胜客最黑暗的料理来了
- “模型”数之联发表通用深度视觉模型可解释性工具,助力破解人工智能模型“黑箱”
- “分子”《科学》(20231013出版)一周论文导读
- “论文”ControlNet、「分割一切」等热门论文获奖,ICCV 2023论文奖项公布
- “蛋白质”利用进化扩散进行蛋白生成,微软开源新型蛋白质生成AI框架EvoDiff
- “样本”存放几十年老样本重见天日:科学家研发单细胞DNA测序法,可用于研究存档的癌症样本
- “氨基酸”南京大学研究团队实现纳米孔蛋白质组学重大突破
- “模型”生成式AI时代的模型压缩与加速,韩松主讲MIT课程,资料全公开
- “锂电池”锂电池回收有了绿色高效新技术
- “结构”解锁蛋白质科学新领域:北大团队提出拓扑改造新方法,助力迈向高分子化学“圣杯”
- “互联网”国家互联网信息办公室关于发布第二批深度合成服务算法备案信息的公告









