“机器”复旦大学、中国科学院团队综述,化学机器学习:基础知识和应用
今天,很高兴为大家分享来自机器之心Pro的复旦大学、中国科学院团队综述,化学机器学习:基础知识和应用,如果您对复旦大学、中国科学院团队综述,化学机器学习:基础知识和应用感兴趣,请往下看。
第一时间掌握
 编辑 | 紫罗
编辑 | 紫罗在过去的十年里,机器学习和人工智能取得了长足的进步,使我们距离智能机器的实现更近了一步。深度学习方法和增强的数据存储能力的在这一进步中发挥了关键作用。机器学习已经在图像和语音识别等领域取得了成功,现在它在以复杂数据和多样化有机分子为特征的化学领域受到了广泛关注。
然而,由于化学家不熟悉现代机器学习算法,他们在采用机器学习应用时经常面临挑战。化学数据集通常表现出对成功实验的偏见,而平衡的视角需要包含成功和失败的实验。此外,文献中合成条件的不完整记录也带来了挑战。
计算化学可以通过量子力学计算构建数据集,因此更容易接受机器学习应用。尽管如此,化学家需要对机器学习有基本的了解,才能利用数据记录和机器学习引导实验的潜力。
近日,复旦大学、中国科学院和贝尔法斯特女王大学(Queen's University Belfast)的研究人员在《Engineering》上发表综述文章:《Machine Learning for Chemistry: Basics and Applications》。
该综述介绍了机器学习的基本组成部分,包括数据库、特征和算法,并重点介绍了机器学习技术在化学领域取得的一些重要成就。综述旨在弥合化学家和现代机器学习算法之间的差距,深入了解机器学习在彻底改变化学研究方面的潜力。

论文链接:https://doi.org/10.1016/j.eng.2023.04.013
综述分为以下几大部分:
首先介绍了流行的化学数据库,它为实践机器学习模型提供了基础。
其次,提出了一些广泛使用的二维 (2D) 和三维 (3D) 特征,这些特征将分子结构转换为机器学习模型可接受的输入。
第三,简要概述了流行的机器学习算法,重点介绍了它们的基本理论框架和适合的应用场景。
第四,更详细地描述了机器学习领域取得重要进展的三个化学领域,包括有机化学中的逆合成、基于机器学习势的原子模拟和多相催化机器学习。
最后,对未来的机器学习应用进行了展望。
ML 中常用化学数据库
没有数据就没有 AI。因此,数据的可用性是现代机器学习应用的先决条件,其中数据集的大小和质量都很重要。在化学领域,收集和编译数据的传统由来已久,数据范围从元素原子光谱到材料宏观特性。化学中的数据科学创造了化学信息学学科,这进一步大大有利于机器学习在化学中的应用。
事实上,尽管从头开始构建大型数据集似乎令人畏惧,但许多化学数据库早在机器学习时代之前就已经可用。表 1 列出了化学领域比较流行的数据库,其中许多数据库都有悠久的数据收集和编译历史。这些数据的来源包括开放专利和研究文章、针对特定属性的高通量实验以及通常基于密度泛函理论 (DFT) 的 QM 计算。
表 1:ML 中常用的流行化学数据库列表。(来源:论文)

2D 和 3D 特征
数据和特征决定了 ML 模型的上限。从源数据预处理得到的特征(通常也称为表示或描述符)是 ML 模型的输入。重要特征的选择(称为特征工程)曾经是 ML 模型训练中最耗时、最费力的工作。虽然深度学习技术可以让机器学习模型学习如何提取特征本身,但它们通常需要相对较大的训练数据集和模型参数空间;因此,它们的计算成本较高,最终创建的机器学习模型可解释性较差。在化学中,不同机器学习模型的输入特征可能不同,但分子/晶体结构表示是特征工程的一般任务。由于关于该主题的优秀评论文章已经发表,这里仅简要介绍与 4 个 ML 模型、5 个应用程序中提到的应用程序相关的一些内容。
分子描述符基本上有两类——即 2D 和 3D 特征。2D 特征关注分子中的键合模式,而忽略空间构象。这些特征源自分子图(以原子为节点,以键为边)或邻接矩阵(即键矩阵)。例如,SMILES 使用人类可读的字符串(例如,乙醇的「CCO」)描述饱和分子,IUPAC 的国际化学标识符 (InChI) 使用严格唯一但不太人类可读的字符串来表示化合物。除了字符串之外,分子的拓扑结构也可以抽象为浮点数的向量。使用 Morgan 算法开发的扩展连接指纹(ECFP),迭代地搜索分子中的子结构并将它们编码为哈希值。
3D 特征是从原子坐标编码的,由于缺乏排列、平移和旋转不变性,原子坐标很难成为 ML 模型的直接输入。优雅的方法旨在保持排列、平移和旋转不变性,并敏感地区分 3D 中的不同结构。这些方法通常基于从原子间距离和原子间角度导出的数值函数,例如最小埋藏体积百分比、原子中心对称函数(ACSF)、Steinhardt 型有序参数,以及功率类型结构描述符(PTSD)。其他方法基于原子密度类似函数,包括但不限于平均空间占据(ASO)、原子位置平滑重叠(SOAP)和基于高斯型轨道的密度向量。
流行的 ML 模型
在特征将数据编码为机器可读的输入后,机器学习模型将输入转换为输出,即预测的属性。机器学习模型不是从理论推导出物理定律,而是在与数据集生成方式相关的易于访问的变量和相关属性之间建立数值联系,而这些属性通常过于复杂而无法通过理论解决。
从广义上讲,机器学习算法(取决于数据集的学习方式)可以分为三大类:用于拟合标记数据的监督学习、用于对未标记数据进行分类的无监督学习以及利用奖励机制来指导数据学习的强化学习 。其中,监督学习由于其对特定目标具有更好的数值可预测性,在科学研究中应用最广泛。尽管 ML 有很多秘诀和类别,但在实践中实现 ML 并不困难,这要归功于许多公开可用的软件包,例如 scikit-learn、PyTorch 和 TensorFlow。
接下来,研究人员介绍了六种常用的机器学习算法:决策树、 前馈神经网络、卷积神经网络、循环神经网络、图神经网络和 Transformer 神经网络。
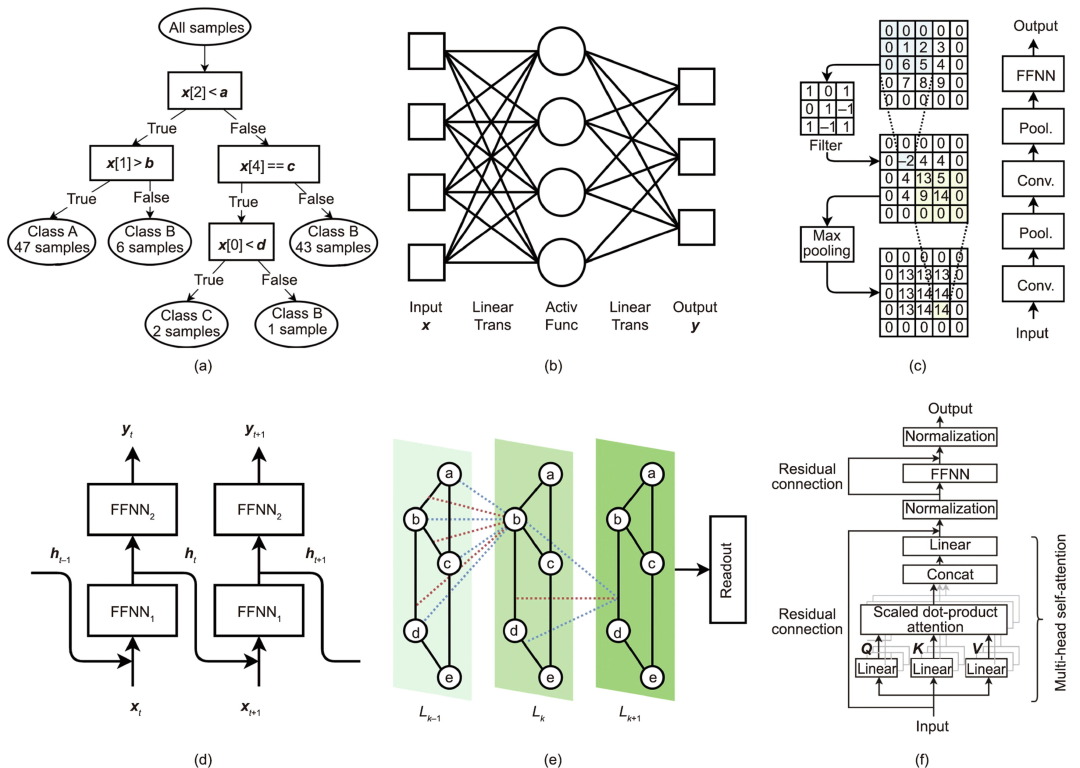 图 1:六种流行的机器学习模型。(来源:论文)
图 1:六种流行的机器学习模型。(来源:论文)ML 在化学中的的应用
在这里,列了 ML 的一些重要应用,以说明如何使用这些 ML 技术来解决化学问题,包括有机化学中的逆合成、计算化学中的 ML 势能以及物理化学中的多相催化。表 2 总结了一些相关文献,其中列出了有关 ML 任务、输入数据、特征、ML 模型和预测目标的信息。
表 2:机器学习在逆合成、机器学习势能和多相催化中的应用总结。(来源:论文)

逆合成
合成计划,也称为逆合成,是化学的核心,回答了如何从现有材料合成所需化合物的问题。在其悠久的历史中,这项任务在很大程度上依赖于经验丰富的化学家的知识。
因此,早在 20 世纪 60 年代 Corey 等人提出的计算机辅助合成计划(CASP)一直是化学领域的热门话题。此后,许多成功的 CAS P程序被开发出来。
由于有机反应丰富且此类数据库相对容易访问,多年来逆合成得到了积极发展,特别是在过去十年中在机器学习技术的帮助下。
反应预测和逆合成是 CASP 中的两个关键模块。反应预测可以分为两类:基于模板的方法和无模板的方法。前者需要一个先验模板库,该模板库可以由专家使用化学信息学进行编码,也可以通过最近流行的原子映射算法从反应数据库中提取。无模板方法通常侧重于预测分子中的反应中心,从而识别最适合连接(断开)的键。
在基于模板的方法中,一种反应物通常会产生太多可能的产物,从而产生过多的候选反应。值得一提的是,基于模板的方法在 CASP 中已经比较成熟,关注点主要包括预测的相关性和模板库的范围。ML 模型的训练中通常必须排除稀有模板。

图 2:(a) 基于模板的反应预测的神经符号方法概述;(b) 用于无模板反应预测的 Seq2seq 模型架构;(c) 指导逆合成的 SCScore 模型方案;(d) MCTS 算法说明。(来源:论文)
近年来出现的无模板方法由于质量和完整性而有可能打破基于模板的方法的局限性。
逆合成更为复杂,因为它的目的是提供一条全局最优的合成途径,这并不像连接最佳的一步反应或选择最短路线那么简单。
尽管目前已经有很多成功的研究,但天然产物的合成仍然是一个挑战。除了复杂分子训练数据的稀疏性之外,大多数模型中通常缺少对映体的定量产率,但这对于正确评估合成路线非常重要。
机器学习势能
机器学习在化学中的另一个重要应用与复杂系统的原子模拟有关,其中机器学习势取代了计算要求较高的 QM 计算来评估 PES。由于 ML 势是在 QM 计算的数据集上进行训练的,因此 ML 势计算可以达到与 QM 相当的精度,但速度要快几个数量级。因此,ML 势方法显著地将原子模拟的领域扩展到具有数千个原子的多元素系统,这可能只能通过传统的经验力场来模拟,尽管力场的可用性高度限制于具有相对简单的 PES 的系统。
自 1995 年第一个 ML 势出现以来,人们提出了许多不同类型的 ML 模型,以及两类 ML 架构(表 2),即 NN 势 和基于 kernel 的势是最受欢迎的。尽管基于 kernel 的势,其超参数比神经网络势要少得多, 它们的计算速度受到训练样本大小的限制。因此,使用基于 kernel 的势来超越大型训练集本质上是困难的,它们更适合单元素系统,例如碳和硅。因此,NN 势正在成为 ML 势计算的主流。
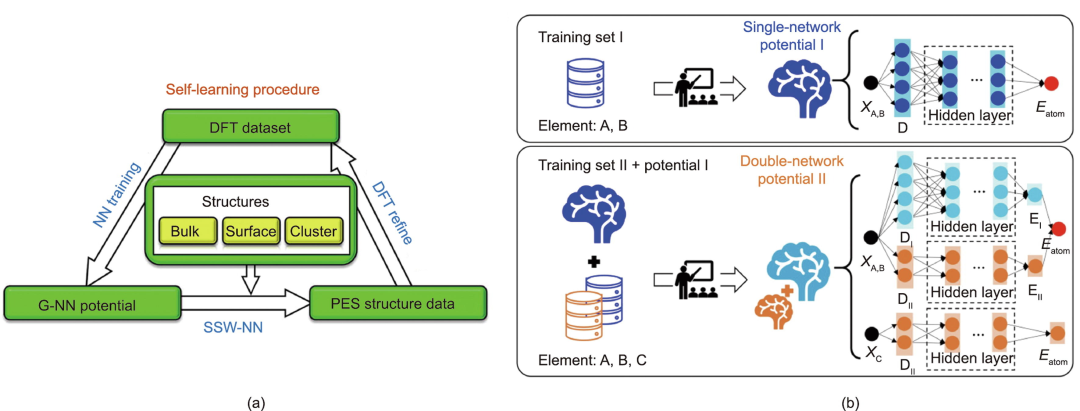 图 3:(a) G-NN 势的 SSW-NN 自学习过程方案。(b) LASP 中实施的双网络框架方案。(来源:论文)
图 3:(a) G-NN 势的 SSW-NN 自学习过程方案。(b) LASP 中实施的双网络框架方案。(来源:论文)用于多相催化的机器学习
由于催化剂结构的复杂性和催化剂在工业中的重要意义,多相催化一直是新技术的主要试验场。早期的机器学习应用可以追溯到 20 世纪 90 年代,通常处于现象学层面,使用简单的机器学习模型学习实验数据来优化催化剂合成和反应条件。这些机器学习应用似乎受到实验数据集可用性的限制,并且由于缺乏基础理解,很可能忽略了实验中隐藏的关键变量,导致机器学习模型的失败。
随着深度学习和机器学习方法的出现,出现了许多更令人兴奋的应用场景,例如机器学习辅助文献分析和人工智能机器人 。
机器学习辅助文献分析利用自然语言处理模型的数据挖掘能力,从文献中提取实验数据。进一步的数据分析将有助于揭示不同实验之间的关键秘诀。
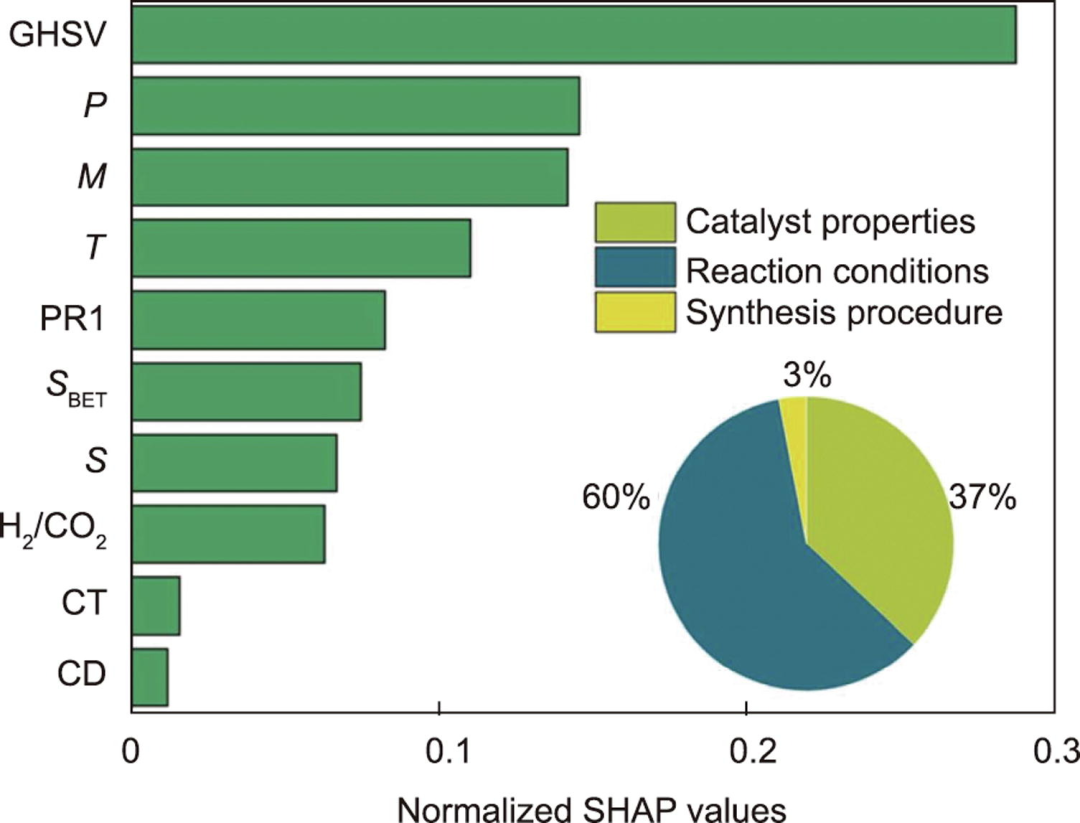 图 4:CO2 加氢制甲醇的特征重要性分析。(来源:论文)
图 4:CO2 加氢制甲醇的特征重要性分析。(来源:论文)化学家机器人被认为是化学的未来,因为它们将自动高效地进行实验,同时保持实验之间最大的数据一致性。
从理论角度来看,机器学习模型还可以用于学习低成本的可计算量,例如分子的吸附能和电子能带结构,这些对于催化很重要 。
另一方面,机器学习原子模拟可以提供有关催化剂结构和反应机理的原子级知识,这有利于催化剂的合理设计。
未来展望
该综述总结了最近化学领域机器学习应用的关键要素,从流行的数据库到常见特征、现代机器学习模型和标准应用场景。
随着最近机器学习应用的成功,我们必须认识到机器学习在化学中的使用带来了许多挑战。例如,一个主要障碍是缺乏高质量的数据,特别是涉及实验的数据。即使有了高通量的实验技术和实验机器人,化学中仍有许多领域必须由人类来产生实验数据。此外,化学家通常不熟悉最先进的机器学习方法和相关计算机科学技术,这导致难以为目标应用设计适当的功能。如何自动提取不同化学问题的特征仍然具有挑战性。最后,大多数基于 FFNN 的机器学习研究很难解释,因此很难转移到新的化学问题。
随着计算设施的快速更新和新的机器学习算法的发展,可以预见更多令人兴奋的机器学习应用即将到来,化学研究的未来必将在机器学习时代被重塑。
虽然未来很难预测,特别是在这样一个快速发展的领域,但毫无疑问,机器学习模型的发展将带来更好的可访问性、更通用性、更好的准确性、更智能,从而提高生产力。机器学习模型与互联网的集成是在世界范围内共享机器学习预测的好方法。
由于元素类型众多、材料复杂性高,化学中机器学习模型的可迁移性是一个常见问题。预测通常必须限于应用的数据库,这只是广阔的化学空间中的本地数据集。预测的准确性迅速下降超出数据集。随着新技术的出现,这个问题可能会得到解决,或者使用更好的机器学习模型,可以学习具有大量拟合参数的更复杂的系统。事实上,数据科学家举办了各种各样的机器学习竞赛,比如 Kaggle,导致了许多优秀算法的诞生。在这方面,化学问题的公开 ML 竞赛仍然有限,需要付出更多努力来促进该领域年轻人才的成长。
对于更智能的机器学习应用,端到端学习是一个有前途的方向,因为它从原始输入而不是手动设计的描述符生成最终输出。这些先进的机器学习模型还应该有助于构建更智能的实验机器人来执行高通量实验。
参考内容:https://phys.org/news/2023-09-machine-chemistry-basics-applications.html
好了,关于复旦大学、中国科学院团队综述,化学机器学习:基础知识和应用就讲到这。
版权及免责声明:凡本网所属版权作品,转载时须获得授权并注明来源“科技金融网”,违者本网将保留追究其相关法律责任的权力。凡转载文章,不代表本网观点和立场,如有侵权,请联系我们删除。
相关文章
- “小行星”我国2030年前后 实现载人登月
- “都是”A股的觉醒之年!
- “孩子”一生的功课
- “益康”倍益康上市几个月收入净利都大减 市值仅几亿 创始人张文有啥办法?
- “科幻”嘉宾共话科幻的未来:被视为“珍贵市场”,中国科幻正青春
- “灯会”红星观察|自贡灯会走出“春节舒适区”:首次试水中秋国庆主题灯会火出圈背后
- “华为”新麒麟全面替代!曝华为正在清理骁龙机型库存:掀起全线新品的“洪流”
- “鸟类”评论丨大楼玻璃贴膜防鸟撞,城市的天空如何助鸟自由飞翔?
- “同济大学”四川“无臂青年”彭超参与杭州亚残运会火炬传递,曾用脚写字考上同济大学研究生
- “模型”人工智能公司OpenCSG发布大模型开源生态社区“传神”
- “模型”解决大模型复现难、协作难, 这支95后学生团队打造了一个国产AI开源社区
- “模型”人工智能公司OpenCSG发布大模型开源生态社区“传神”
- “架构”清华朱文武团队:开源世界首个轻量图自动机器学习库AutoGL-light
- “液态”我国学者构建液态金属磁性微型软体机器人,可用于临床医学
- “模型”北理工团队在人工智能图像识别领域取得新进展
- “人类”有了GPT-4之后,机器人把转笔、盘核桃都学会了
- “模型”参数少近一半,性能逼近谷歌Minerva,又一个数学大模型开源了
- “模型”评论能力强于GPT-4,上交开源13B评估大模型Auto-J
- “模型”端侧首次实现70亿AI语言大模型 联发科vivo强强联手
- “产品”用12万UGC造一个爆款 可以科技如何扎根家庭机器人赛道?









